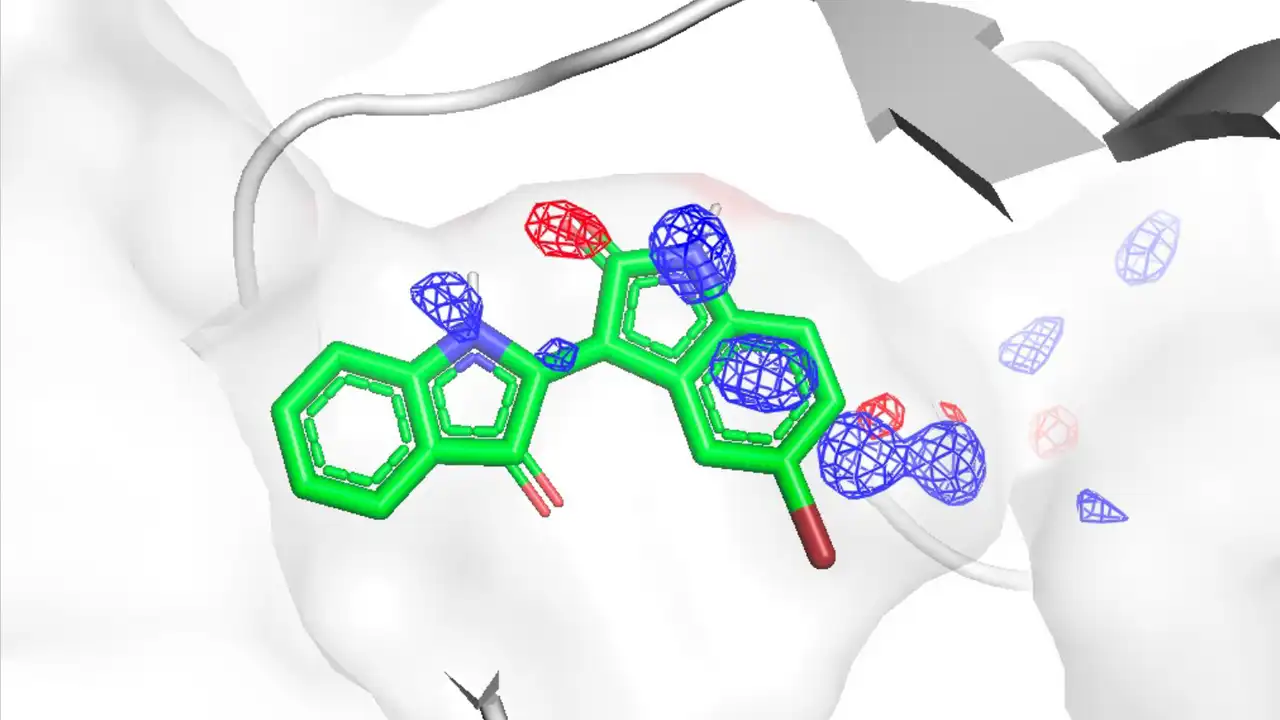
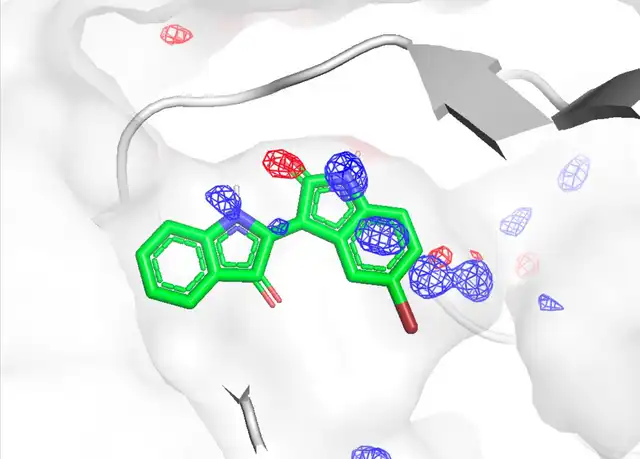
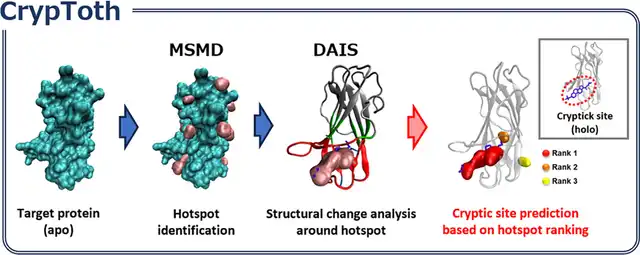
Jun Koseki, Chie Motono, Keisuke Yanagisawa, Genki Kudo, Ryunosuke Yoshino, Takatsugu Hirokawa, Kenichiro Imai, "CrypToth: Cryptic Pocket Detection through Mixed-Solvent Molecular Dynamics Simulations-Based Topological Data Analysis", Journal of Chemical Information and Modeling 65, 5567-5575, 2025/5. DOI: 10.1021/acs.jcim.4c02111
Genki Kudo, Keisuke Yanagisawa, Ryunosuke Yoshino, Takatsugu Hirokawa, "AAp-MSMD: Amino Acid Preference Mapping on Protein–Protein Interaction Surfaces Using Mixed-Solvent Molecular Dynamics", Journal of Chemical Information and Modeling 63, 7768-7777, 2023/12. DOI: 10.1021/acs.jcim.3c01677
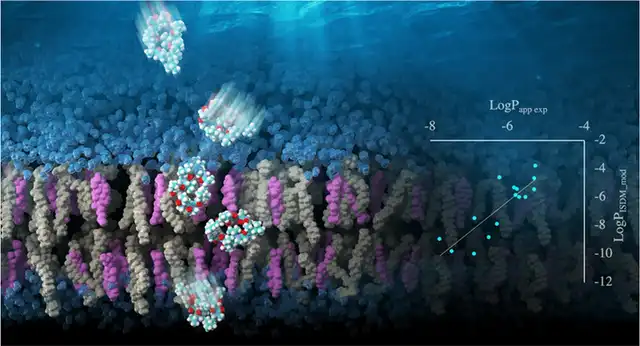
Masatake Sugita, Takuya Fujie, Keisuke Yanagisawa, Masahito Ohue, Yutaka Akiyama, "Lipid Composition Is Critical for Accurate Membrane Permeability Prediction of Cyclic Peptides by Molecular Dynamics Simulations", Journal of Chemical Information and Modeling 62, 4549-4560, 2022/9. DOI: 10.1021/acs.jcim.2c00931
Keisuke Yanagisawa, Yoshitaka Moriwaki, Tohru Terada, Kentaro Shimizu, "EXPRORER: Rational Cosolvent Set Construction Method for Cosolvent Molecular Dynamics Using Large-Scale Computation", Journal of Chemical Information and Modeling 61, 2744-2753, 2021/6. DOI: 10.1021/acs.jcim.1c00134