Molecular Dynamics (MD) Simulation
Molecular dynamics (MD) simulation computes the motions of atoms and molecules based on physical laws, enabling all-atom analysis of the dynamic behavior of biomolecules. Our lab conducts protein-compound interaction analysis using the mixed-solvent MD (MSMD) method, which mixes probe molecules (co-solvent molecules) into water, as well as highly accurate cyclic peptide membrane permeability prediction via massively large-scale computations using enhanced sampling methods.
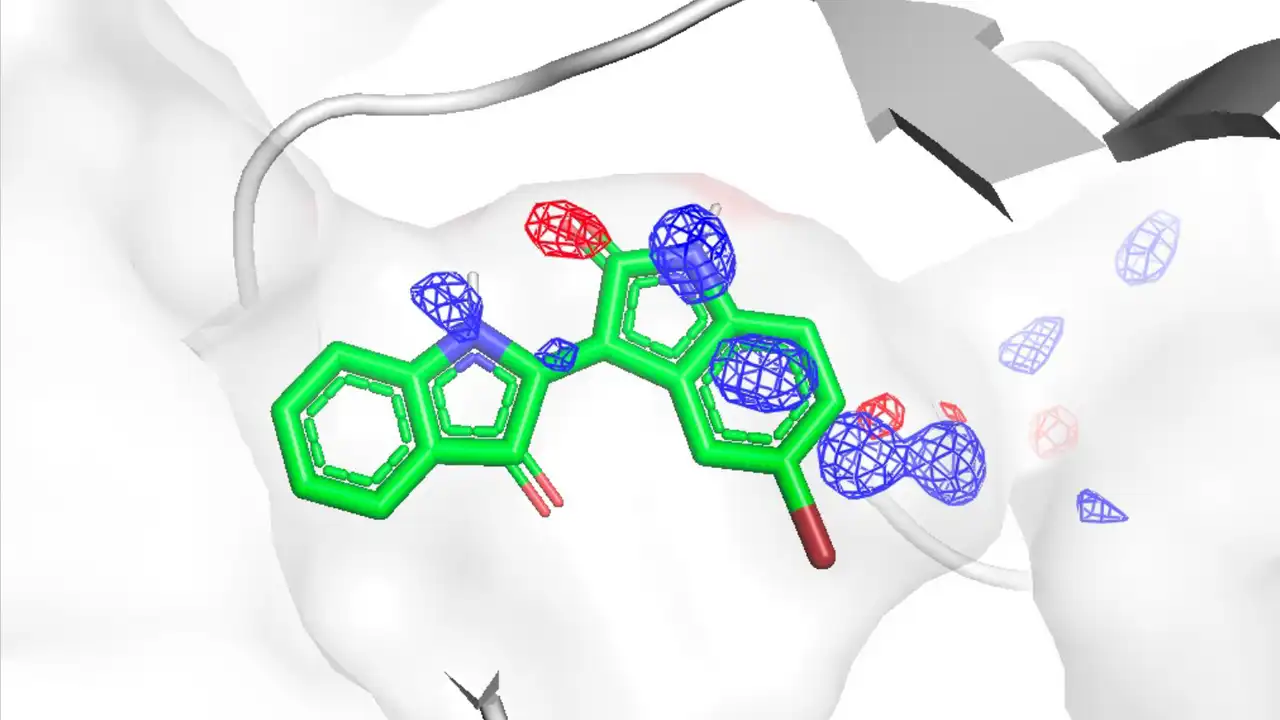
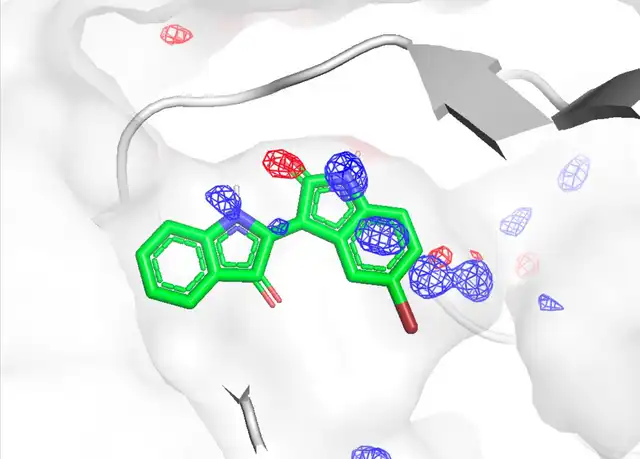
- Binding site prediction: Prediction of compound and peptide binding sites using spatial probability distribution maps (PMAPs) of probe molecules
- Improvement of docking calculations with MD: Application of grid free energy (GFE) obtained from MSMD simulations to docking calculation scoring
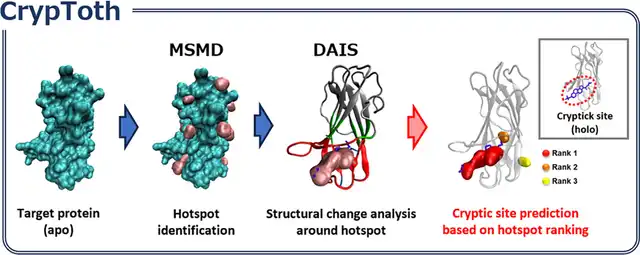
- Cryptic binding site prediction: Prediction of hidden binding sites (cryptic sites) that can only be observed through protein conformational changes
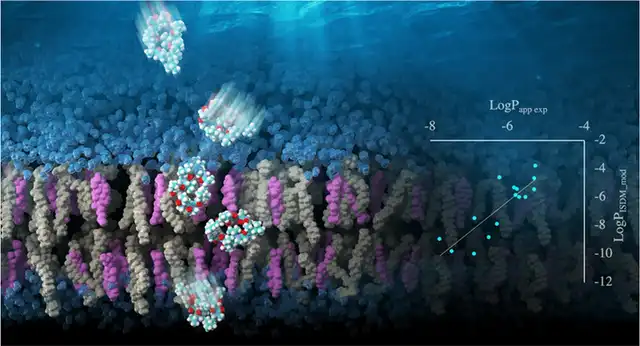
- Membrane permeability prediction for cyclic peptides: Development of highly accurate membrane permeability prediction methods using REST/REUS MD simulations